

Памятка предназначена для субъектов обращения лекарственных средств, изделий медицинского назначения и медицинской техники
03.04.2017по организации оценки условий производства, системы обеспечения качества и (или) аналитической экспертизы (в условиях производства) при государственной регистрации лекарственных средств, изделий медицинского назначения, медицинской техники и оценки безопасности и качества лекарственных средств и изделий медицинского назначения, зарегистрированных в Республике Казахстан
1. Введение
1.1 Настоящая Памятка предназначена для субъектов обращения лекарственных средств, изделий медицинского назначения и медицинской техники (далее – ЛС, ИМН и МТ), описывает процесс организации и проведения оценки условий производства и системы обеспечения качества, аналитической экспертизы (в условиях производства) при государственной регистрации ЛС, ИМН и МТ, а также при проведении оценки безопасности и качества лекарственных средств и изделий медицинского назначения, зарегистрированных в Республике Казахстан (далее – оценка условий производства).
1.2 Настоящая Памятка разработана в соответствии с пунктом 11 Статьи 71 Кодекса Республики Казахстан от 18 сентября 2009 года № 193-IV «О здоровье народа и системе здравоохранения», приказом Министра здравоохранения Республики Казахстан от 19 ноября 2009 года № 743 «Об утверждении Правил оценки условий производства и системы обеспечения качества при государственной регистрации лекарственных средств, изделия медицинского назначения и медицинской техники», приказом Министра здравоохранения Республики Казахстан от 18 ноября 2009 года № 736 «Об утверждении Правил проведения экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники», приказом Министра здравоохранения и социального развития Республики Казахстан от 26 ноября 2014 года № 269 и внутренними регламентами РГП на ПХВ «Национальный Центр экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники» Министерства здравоохранения Республики Казахстан (далее – РГП на ПХВ «НЦЭЛС, ИМН и МТ» МЗ РК).
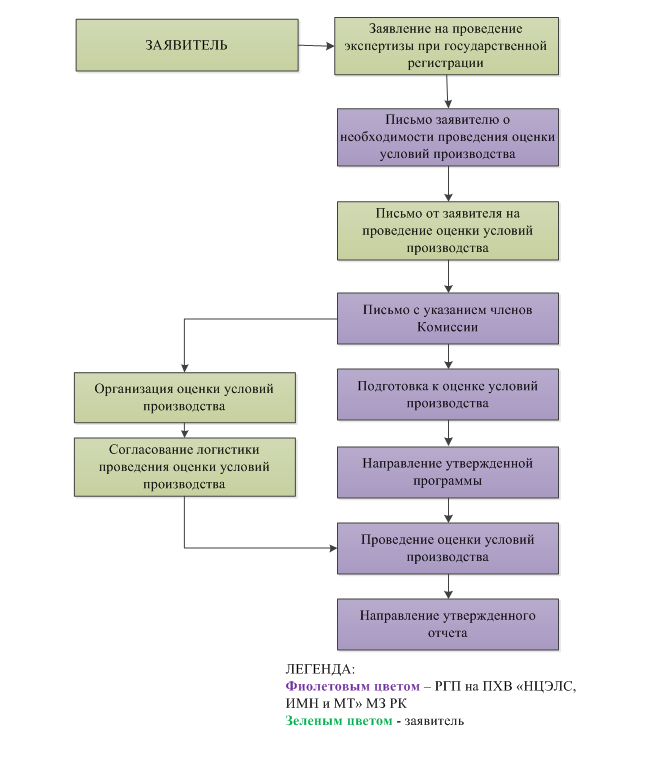
Блок-схема процесса организации и проведения оценки условий производства представлена в Приложении 1.
2. Основание для проведения оценки условий производства
Оценка условий производства при государственной регистрации ЛС, ИМН и МТ осуществляется в случаях:
2.1 регистрации ЛС, ИМН и МТ организацией-производителем, ранее не регистрировавшей продукцию в Республике Казахстан;
2.2 регистрации с производственных участков (площадок), ранее не поставлявших продукцию в Республику Казахстан;
2.3 регистрации оригинального лекарственного средства, впервые поданного на государственную регистрацию;
2.4 отсутствия сертификата «Надлежащей производственной практики (GMP)»;
2.5 невозможности проведения аналитической экспертизы в связи с отсутствием стандартных образцов активных веществ и примесей, образцов лекарственного средства вследствие их труднодоступности и (или) высокой стоимости; штаммов микроорганизмов и биологических культур вследствие их патогенности, токсичности; специального оборудования и расходных материалов, а также невозможности соблюдения условий их транспортирования, поставки на территорию Республики Казахстан и (или) хранения;
2.6 невозможности проведения оценки достоверности данных о безопасности и эффективности ЛС и соответствия условий проведения доклинических и (или) клинических испытаний по документам, представленных в регистрационном досье;
2.7 невозможности оценить надлежащую деятельность службы фармаконадзора производителя по постмаркетинговому контролю безопасности и эффективности лекарственного препарата (фармаконадзор) по документам, предоставленных в регистрационном досье.
3. Цели оценки условий производства
3.1 Целью оценки условий производства ЛС является подтверждение соответствия производства, производственного участка (площадки) требованиям «Надлежащей производственной практики (GMP)».
3.2 Целью оценки условий производства ИМН и МТ является подтверждение соответствия производства, производственного участка (площадки) требованиям «Надлежащей производственной практики (GMP)» и (или) межгосударственного стандарта ISO 13485 «Изделия медицинские. Системы менеджмента качества. Системные требования для целей регулирования» (или его более новая редакция).
3.3 Целью аналитической экспертизы в условиях производства является подтверждение соответствия образцов ЛС, ИМН требованиям нормативного документа по контролю качества и безопасности по результатам физических, химических, физико-химических и биологических испытаний, проведенных в условиях производства, а также проведение оценки нормативного документа по контролю качества и безопасности на предмет воспроизводимости методик анализа.
4. Инициирование проведения оценки условий производства
4.1 Решение о проведении оценки условий производства может быть принято на любом этапе проведения экспертных работ при государственной регистрации лекарственных средств, изделий медицинского назначения и медицинской техники.
4.2 Решение о проведении оценки условий производства принимает государственный орган в сфере обращения ЛС, ИМН и МТ (далее – государственный орган) по представлению РГП на ПХВ «НЦЭЛС, ИМН и МТ» МЗ РК на любом этапе проведения экспертных работ в порядке, регулирующем процедуру государственной регистрации ЛС, ИМН и МТ в Республике Казахстан.
5. Проведение оценки условий производства
5.1 Оценка условий производства при государственной регистрации лекарственных средств, изделий медицинского назначения и медицинской техники осуществляется Комиссией (далее – Комиссия), из числа представителей государственного органа и специалистов РГП на ПХВ «НЦЭЛС, ИМН и МТ» МЗ РК, не принимающих непосредственного участия в разработке и производстве лекарственного средства, изделия медицинского назначения и медицинской техники путем посещения субъекта по производству лекарственных средств, изделий медицинского назначения и медицинской техники (далее – организация-производитель) за счет его средств.
5.2 Состав Комиссии для проведения оценки условий производства одной производственной площадки может включать до 5-ти экспертов, в том числе внешние эксперты, в зависимости от типа оценки производства, объема работ, специфики лекарственного средства, изделия медицинского назначения и медицинской техники.
5.3 Комиссию возглавляет руководитель, который несет ответственность за подготовку программы, координацию деятельности членов Комиссии, проведение оценки условий производства, классификацию выявленных несоответствий и написание отчета.
6. Информирование заявителя о необходимости проведения оценки условий производства
После инициации проведения оценки условий производства и принятия решения государственным органом о посещении организации-производителя, РГП на ПХВ «НЦЭЛС, ИМН и МТ» МЗ РК направляется заявителю уведомление о необходимости проведения оценки условий производства.
Примечание Заявитель – разработчик, организация-производитель, держатель регистрационного удостоверения или их доверенное лицо, уполномоченное подавать заявление, документы и материалы на проведение экспертизы ЛС, ИМН и МТ для их государственной регистрации, перерегистрации, внесения изменений в регистрационное досье.
7. Заявление на проведение оценки условий производства
7.1 После получения уведомления о необходимости проведения оценки условий производства, заявитель направляет заявление в РГП на ПХВ «НЦЭЛС, ИМН и МТ» МЗ РК на проведение оценки условий производства по форме согласно Приложению 2.
7.2 В случае обращения заявителя о проведении аналитической экспертизы в условиях производства до начала проведения работ по экспертизе при государственной регистрации он заполняет и направляет в РГП на ПХВ «НЦЭЛС, ИМН и МТ» МЗ РК заявление по форме согласно Приложению 3.
7.3 При проведении оценки условий производства производственной площадки завода-производителя или контрактной площадки завода-производителя дополнительно к заявлению предоставляется Досье производственной площадки (с переводом на русский язык) со всеми приложениями.
7.4 При отсутствии электронной версии документов, заявитель предоставляет по одной бумажной копии документов каждому эксперту.
8. Организация проведения оценки условий производства
Для проведения оценки условий производства заявитель назначает ответственное лицо от организации-производителя, которое:
1) определяет контакты и средства связи с РГП на ПХВ «НЦЭЛС, ИМН и МТ» МЗ РК;
2) обеспечивает перевод необходимой информации на русский язык, а также условия работы Комиссии.
3) заблаговременно согласовывает с руководителем Комиссии и начальником Управления по внедрению и развитию надлежащих фармацевтических практик и международных стандартов (далее - Управление) сроки проведения оценки условий производства;
4) бронирует места проживания, билеты на международный и внутренний транспорт (авиа, железнодорожный, автомобильный) по согласованию с руководителем Комиссии;
5) обеспечивает своевременное оформление виз и страховых полисов;
6) своевременно предоставляет ответы на корреспонденцию, поступающую от РГП на ПХВ «НЦЭЛС, ИМН и МТ» МЗ РК;
7) предоставляет в Управление в сканированном электронном варианте перевод программы на английский и (или) иной язык(и), который(ые) был(и) направлен(ы) в организацию-производителя.
Подтверждающие документы должны быть предоставлены не менее, чем за 1 (одну) неделю до начала проведения оценки условий производства и (или) аналитической экспертизы в Управление для формирования служебной записки о командировании руководителя и членов Комиссии.
В случае получения визы руководителя и членов Комиссии, сроки предоставления документов корректируются с учетом получения виз.
9. Подготовка к проведению оценки условий производства
После согласования сроков проведения оценки условий производства, руководитель Комиссии совместно с членами формирует программу оценки условий производства, согласовывает и утверждает ее в соответствии с внутренним регламентом РГП на ПХВ «НЦЭЛС, ИМН и МТ» МЗ РК, после чего направляет заявителю.
10. Проведение оценки условий производства
10.1 Рабочим языком проведения оценки условий производства является русский. При необходимости заявитель обеспечивает качественный, синхронный перевод во время переписки и проведения оценки условий производства на русский язык. Расходы, связанные с переводом, оплачивает заявитель.
Перевод документов и устный перевод во время проведения оценки условий производства должен быть профессиональным, максимально соответствующим оригинальному тексту и информации, предоставленной организацией-производителем. Потребность в переводчике определяется руководителем Комиссии.
10.2 В процессе проведения оценки условий производства члены Комиссии могут снимать копии с документов, необходимых для составления отчета о проведении оценки условий производства при государственной регистрации лекарственных средств, изделий медицинского назначения и медицинской техники.
10.3 Члены Комиссии соблюдают конфиденциальность сведений, получаемых в процессе подготовки и проведения оценки условий производства, а также результатов оценки условий производства.
10.4 Организация-производитель назначает ответственное лицо для сопровождения и работы с членами Комиссии при проведении оценки условий производства.
Сопровождающие членов Комиссии лица, должны присутствовать при работе Комиссии, но не должны оказывать влияние или вмешиваться в проведение оценки условий производства.
Важно!
На момент проведения оценки условий производства сертификат(ы)
GMP и (или) ISO должны быть актуальными (действующими).
11. Сроки и условия проведения оценки условий производства
11.1 Заявитель организовывает проведение оценки условий производства в течение тридцати календарных дней после получения им письма от РГП на ПХВ «НЦЭЛС, ИМН и МТ» МЗ РК о необходимости проведения оценки условий производства, но не более чем через девяносто календарных дней после получения информации.
11.2 Оценка условий производства осуществляется в течение 2-5 рабочих дней на одной производственной площадке. Сроки проведения аналитической экспертизы в условиях производства устанавливаются в зависимости от сроков, предусмотренных методиками испытаний в нормативных документах.
11.3 Отчет о проведении оценки производства составляется в течение 30 календарных дней с момента окончания оценки производства в трех экземплярах, из которых: первый - направляется в государственный орган, второй остается в РГП на ПХВ «НЦЭЛС, ИМН и МТ» МЗ РК, третий - в организацию-производителя ЛС, ИМН и МТ.
11.4 Отчет о проведении аналитической экспертизы с выездом в лабораторию завода-производителя и (или) контрактную лабораторию предоставляется в течение 30 календарных дней с момента окончания проведения аналитической экспертизы в трех экземплярах, из которых: первый - направляется в государственный орган, второй остается в РГП на ПХВ «НЦЭЛС, ИМН и МТ» МЗ РК, третий - в организацию-производителя ЛС, ИМН и МТ. Если аналитическими методиками предусмотрен более длительный срок проведения испытаний, срок предоставления отчета продлевается на время предоставления результатов данных испытаний.
12. Командировки, логистика, оплата
12.1 Заявитель оплачивает все расходы, связанные с проведением оценки условий производства и (или) аналитической экспертизы на производстве, которые включают: проезд (в оба конца), проезд внутри населенного пункта (связанного с местом работы, размещением в гостинице, аэропортом (железнодорожным) вокзалом (автовокзалом)); проживание; питание, в том числе во время в пути; визовые и страховые расходы.
12.2 Обязательное наличие страхового полиса для руководителя и членов Комиссии на время пребывания в командировке для проведения оценки условий производства.
12.3 В случае оценки условий производства, связанной с посещением нескольких объектов, трансфер от одного объекта до другого объекта обеспечивается заявителем.
Приложение 1
Блок-схема процесса организации и проведения оценки условий производства
Приложение 2
Заявление на проведение оценки условий производства и системы обеспечения качества при государственной регистрации лекарственного средства, изделия медицинского назначения, медицинской техники и оценки безопасности и качества лекарственных средств и изделий медицинского назначения, зарегистрированных в Республике Казахстан
(наименование и адрес заявителя)
в лице _________________________________________________________________
(должность, Ф.И.О.)
просит провести оценку условий производства, системы обеспечения качества и (или) аналитическую экспертизу при государственной регистрации (нужное подчеркнуть) по заявлению № ___________
________________________________________________________________________
(полное наименование заявленной на регистрацию продукции)
________________________________________________________________________
(номер и дата заявления на проведение экспертизы
при государственной регистрации ЛС, ИМН и МТ)
в соответствии с подпунктом (ами) ________ пункта 3 Правил оценки условий производства и системы обеспечения качества при государственной регистрации лекарственных средств, изделий медицинского назначения и медицинской техники, утвержденных приказом МЗ РК от 19.11.2009 г. № 743.
Информация о производителе и производственной(ых) площадке(ах)
|
Тип производителя |
Наименование, страна |
Адрес местонахождения (с указанием GPS расположения объекта) |
Контакты (телефон, е-mail) |
|
1 |
2 |
3 |
4 |
|
Производитель
|
|
|
|
|
Предприятие-упаковщик |
|
|
|
|
Предприятие, осуществляющее выпускающий контроль продукции |
|
|
|
|
Держатель лицензии |
|
|
|
|
Контрактная организация (если применимо) |
|
|
|
Аутентичность перевода Досье производственной площадки на русском языке подтверждаем с его оригинальным документом.
Приложение 1: Досье производственного участка и (или) Руководство по качеству с указанием GPS расположения объекта.
Приложение 2: Договор с контрактной организацией (площадкой) завода-производителя, если на ней осуществляется производство.
Должность _______________ (И. Фамилия)
подпись
Дата
Место печати
Приложение 3
Заявление на проведение аналитической экспертизы
в условиях производства при государственной регистрации лекарственного средства, изделия медицинского назначения, медицинской техники и оценки безопасности и качества лекарственных средств и изделий медицинского назначения, зарегистрированных в Республике Казахстан
(наименование и адрес заявителя)
в лице _________________________________________________________________
(должность, Ф.И.О.)
просит провести оценку условий производства, системы обеспечения качества и (или) аналитическую экспертизу при государственной регистрации (нужное подчеркнуть) по заявлению № ___________
(полное наименование заявленной на регистрацию продукции)
(номер и дата заявления на проведение экспертизы
при государственной регистрации ЛС, ИМН и МТ)
в соответствии с подпунктом (ами) ________ пункта 3 Правил оценки условий производства и системы обеспечения качества при государственной регистрации лекарственных средств, изделий медицинского назначения и медицинской техники, утвержденных приказом МЗ РК от 19.11.2009 г. № 743.
Информация о производителе и производственной(ых) площадке(ах)
|
Тип производителя |
Наименование, страна |
Адрес местонахождения (с указанием GPS расположения объекта) |
Контакты (телефон, е-mail) |
|
1 |
2 |
3 |
4 |
|
Производитель
|
|
|
|
|
Предприятие-упаковщик |
|
|
|
|
Предприятие, осуществляющее выпускающий контроль продукции |
|
|
|
|
Держатель лицензии |
|
|
|
|
Контрактная лаборатория (если применимо) |
|
|
|
Обоснование необходимости проведения аналитической экспертизы в условиях производства
|
Причина |
Обоснование |
Подтверждающий документ |
|
1 |
2 |
3 |
|
Труднодоступность и (или) высокая стоимость образцов лекарственного средства, изделия медицинского назначения |
|
|
|
Отсутствие стандартных образцов активных веществ и примесей |
|
|
|
Отсутствие штаммов микроорганизмов и биологических культур вследствие их патогенности, токсичности |
|
|
|
Отсутствие специального оборудования и расходных материалов |
|
|
|
Невозможность соблюдения условий транспортирования, поставки на территорию Республики Казахстан и (или) хранения образцов лекарственного средства, изделия медицинского назначения, стандартных образцов активных веществ и примесей, расходных материалов |
|
|
Аутентичность перевода Досье производственной площадки, спецификации, АНД на русском языке подтверждаем с его оригинальным документом.
Приложение 1 Спецификация на готовый продукт.
Приложение 2 Договор с контрактной лабораторией, если в ней осуществляется аналитическая экспертиза
Должность _______________ (И. Фамилия)
подпись
Дата
Место печати