

Часто встречающиеся несоответствия в итоговых документах при оформлении проектов приказов
17.04.2013
Лекарственные препараты поступают в обращение с информацией для потребителей, указанной в инструкции по медицинскому применению, и с соответствующей маркировкой упаковки.
Маркировка потребительской упаковки должна соответствовать нормативно-техническому документу по контролю за качеством и безопасностью лекарственного средства (далее АНД (ВАНД) и инструкции по медицинскому применению, утверждаемых при государственной регистрации, перерегистрации, внесении изменений в регистрационное досье лекарственного средства.
В 2011-2012 гг. введены в действие нормативные правовые документы, регламентирующие правила оформления проектов инструкций по медицинскому применению, АНД (ВАНД) и маркировки упаковок:
· приказ Министра здравоохранения Республики Казахстан от 8 июня 2011 года №366 «О внесении дополнения в приказ Министра здравоохранения Республики Казахстан от 2 ноября 2009 года № 634 «Об утверждении Правил маркировки лекарственных средств, изделий медицинского назначения и медицинской техники»;
- приказ Министра здравоохранения Республики Казахстан от 28 октября 2011 года №745 «О внесении изменений в приказ Министра здравоохранения Республики Казахстан от 18 ноября 2009 года № 736 «Об утверждении Правил проведения экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники»;
- постановление Правительства Республики Казахстан от 30 декабря 2011 года №1692 «Об утверждении Правил маркировки лекарственных средств, изделий медицинского назначения и медицинской техники и внесении изменений в постановление Правительства Республики Казахстан» от 14 июля 2010 года № 712 «Об утверждении технического регламента «Требования к безопасности лекарственных средств»;
- приказ министра здравоохранения Республики Казахстан от 15 февраля 2012 года №84 «О внесении изменений и дополнений в приказ Министра здравоохранения Республики Казахстан от 18 ноября 2009 года № 735 «Об утверждении Правил государственной регистрации, перерегистрации и внесения изменений в регистрационное досье лекарственного средства, изделий медицинского назначения и медицинской техники» и др.
Экспертиза лекарственных препаратов при государственной регистрации, перерегистрации и внесении изменений в регистрационное досье типа II состоит из четырех этапов:
- первичной экспертизы;
- аналитической экспертизы;
- специализированной фармацевтической экспертизы;
- специализированной фармакологической экспертизы.
При внесении изменений в регистрационное досье типа I – первичной экспертизы и (или) аналитической, фармацевтической и фармакологической экспертиз в соответствии с требованиями вносимых изменений.
После прохождения полного цикла экспертных работ итоговые документы на лекарственные препараты (проекты инструкций по медицинскому применению, АНД (ВАНД) и макеты упаковок) поступают на завершающий этап – «Заключение о безопасности» – в Управление первичной экспертизы лекарственных средств (далее – УПЭЛС).
После укомплектования всех документов специалист УПЭЛС в течение 10 дней оформляет заключение о безопасности, эффективности и качестве лекарственных средств и проект приказа по регистрации, перерегистрации и внесению изменений в регистрационное досье.
Предварительно заявители проверяют размещенные на сайте www.dari.kz сведения по препаратам: торговое название, МНН, лекарственную форму, дозировку, состав, описание, форму выпуска и упаковки, условия и срок хранения, условия отпуска из аптек, наименование производителя на государственном и русском языках и пр.
В случае выявления несоответствий в проектах инструкций по медицинскому применению, АНД (ВАНД) и макетах упаковок заявители с сопроводительным письмом представляют обновленные документы, согласно которым специалист УПЭЛС вносит необходимые коррективы в программу «Экспертиза ЛС, ИМН и МТ» (далее – Программа).
Электронные версии исправленных проектов инструкций по медицинскому применению, АНД (ВАНД) и макетов упаковок в Программу вводят ученые секретари управлений фармакологической и фармацевтической экспертиз, соответственно, бумажные версии вышеуказанных документов передают в УПЭЛС.
При отсутствии любых несоответствий заявители представляют письмо-согласование о достоверности размещенных на сайте сведений.
На основании письма-согласования специалист УПЭЛС оформляет:
- заключение о безопасности, эффективности и качестве лекарственных средств, заявленных на государственную регистрацию и перерегистрацию, внесение изменений в РК;
- перечень лекарственных средств, рекомендованных к государственной регистрации, перерегистрации, внесению изменений и разрешенных к медицинскому применению на территории РК;
- сведения о прохождении экспертных работ при государственной регистрации, перерегистрации лекарственных средств, внесении изменений в регистрационное досье.
Нами проанализированы и систематизированы ошибки, допускаемые заявителями при подготовке проектов инструкций по медицинскому применению, АНД (ВАНД) и макетов упаковок.
В таблице 1 приведены замечания, часто выставляемые специалистами на этапе «Заключение о безопасности», при формировании проекта приказа по регистрации, перерегистрации лекарственных средств и внесению изменений в регистрационное досье.
Таблица 1
|
№ |
Тип документа |
Замечания |
|
1 |
Проект АНД (ВАНД) |
А). Необходимо правильно указывать номер и наименование нормативного аналитического документа, утвержденного при регистрации, взамен которого вводится АНД. |
|
Б). Отечественным производителям при регистрации препарата необходимо оформлять временный аналитический нормативный документ (ВАНД) со сроком действия 3 года. |
||
|
В). Унифицировать общие данные по препарату (лекарственную форму, состав, описание, форму выпуска и упаковки, условия хранения) с проектом инструкции по медицинскому применению и макетами упаковок. |
||
|
2 |
Проект инструкции |
А) унифицировать общие данные по препарату (лекарственную форму, состав, описание, форму выпуска и упаковки, условия хранения) с проектом АНД (ВАНД). |
|
3 |
Макеты упаковок |
А). Необходимо указывать предусмотренные приказами обязательные предупредительные надписи, перечень вспомогательных веществ (антимикробные консерванты, красители, сахара и этанол), для инфузионных растворов – количественный состав вспомогательных веществ. |
|
Б) Не наносить на упаковку сведения рекламного характера. |
||
|
В). Оформлять дизайн упаковки лекарственного препарата одной и той же лекарственной формы, содержащего разные количества активных веществ, в различном цветовом исполнении с указанием цифровой идентификации цветов изображения Pantone. |
||
|
Г). Указывать маркировкy первичной и вторичной упаковок на государственном и русском языках. |
||
|
Д). Согласовывать текст маркировки на государственном языке со специалистами отдела переводов. |
||
|
Е). Представлять бумажные и электронные макеты упаковок, соответствующие описанию в разделе «Упаковка» проекта АНД (ВАНД) |
||
|
Ж). Не указывать на упаковке прежний регистрационный номер в случае изменения статуса заявки «Перерегистрация» на «Регистрация» при подаче препарата на плановую перерегистрацию. |
||
|
И). Необходимо корректно указывать аббревиатуру в номерах регистрационных удостоверений на лекарственные и иммунобиологические препараты: «РК-ЛС-» и «РК-БП-», соответственно |
В таблице 2 представлена информация по срокам оформления проектов приказов по регистрации, перерегистрации и внесению изменений в регистрационное досье за 2012 г.
Таблица 2
|
Тип регистрации |
Количество препаратов, включенных в проект приказа в срок |
Всего |
|
|
до 10 дней |
более 10 дней |
||
|
Регистрация |
714 |
138 |
852 |
|
Перерегистрация |
565 |
151 |
716 |
|
Внесение изменений |
1373 |
572 |
1945 |
|
Всего: |
2652 |
861 |
3513 |
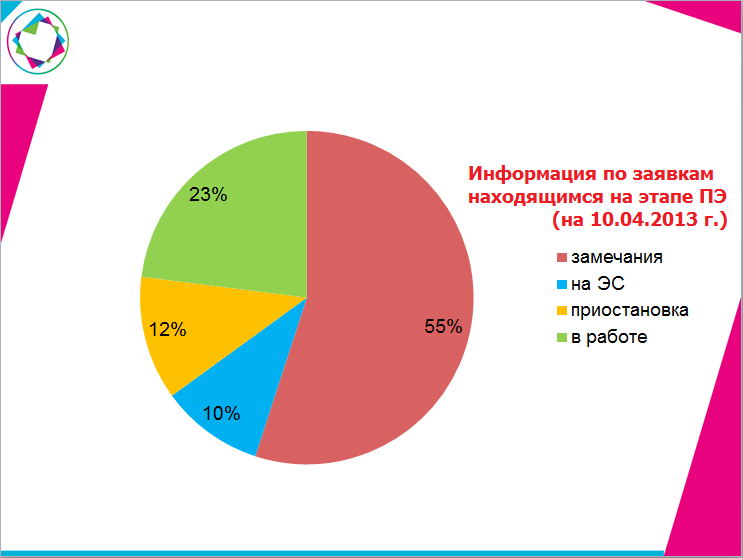
Согласно приведенным в таблице данным, более 20% препаратов находятся на этапе «Заключение о безопасности» дольше регламентированного срока.
Очень часто заявители приостанавливают экспертные работы для согласования несоответствий, выявленных на заключительном этапе, с заводом-изготовителем.
Приостановка работ и последующая переписка сказываются на сроках оформления проектов приказов.
Таким образом, для обеспечения своевременного проведения экспертных работ заявителям необходимо с большей ответственностью относиться к подготовке документов регистрационного досье и своевременно информировать о любых несоответствиях, касающихся сведений о препаратах, размещенных на сайте.
управление первичной экспертизы лекарственных средств,
РГП на ПХВ «Национальный Центр экспертизы лекарственных средств,
изделий медицинского назначения и медицинской техники» МЗ РК, г. Алматы.