

Европейская фармакопея и ее роль в развитии национальных стандартов качества лекарственных средств
01.01.1970Знаковым событием прошедшего года явилось вступление Республики Казахстан в статус страны-наблюдателя в Комиссии Европейской Фармакопеи (Европейская фармакопейная комиссия). Решение было единогласно принято на Июньской сессии комиссии. Ранее в ее состав входило 36 государств-членов, в том числе 24 страны Европейского Союза (ЕС), а статус наблюдателей в ней имело 19 государств. Принятие Республики Казахстан и Российской Федерации наблюдателями, а также переход Польши в статус члена Европейской фармакопейной комиссии изменило представительство государств до 37 и 21 соответственно.
Традиционно сессии Европейской фармакопейной комиссии проходят в г. Страсбурге с регулярностью три раза в год (в марте, июне и ноябре). На Ноябрьской сессии Казахстан представляли генеральный директор РГП «Национальный Центр экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники» Г.Д. Бердимуратова и директор Фармакопейного центра профессор А.У. Тулегенова.
Статус наблюдателя позволит республике решать следующие вопросы:
- привлечение к научным исследованиям, проводимым под эгидой ЕФ;
- приобретение собственного опыта на основании европейского опыта работы в области контроля качества и методов анализа лекарственных средств;
- определение национальных подходов и путей развития в данной области.
Важно, что вступление Казахстана в статус наблюдателя обязывает следовать принципам и подходам Европейской Фармакопеи (ЕФ) в разработке национальных стандартов качества лекарственных средств. Понимание главной цели, фундаментальных принципов и функций ЕФ позволяет определить рациональные пути развития и выработать корректные решения в национальной системе контроля качества лекарственных средств.
Основополагающая цель и задачи ЕФ сформулированы Европейской фармакопейной комиссией в предисловии к 3-му изданию: «Цель Европейской Фармакопеи состоит в том, чтобы способствовать сохранению здоровья населения путем предоставления общепризнанных стандартов для применения их работниками службы здравоохранения и другими лицами, имеющими отношение к качеству лекарственных препаратов. Такие стандарты, будучи основой для безопасности применения лекарственных препаратов потребителями и заказчиками, должны характеризоваться соответствующим качеством. Их наличие облегчает свободное движение лекарственных препаратов на рынках Европы и гарантирует качество лекарственных препаратов, экспортируемых из Европы. Монографии Европейской Фармакопеи и другие положения разработаны с тем, чтобы отвечать потребностям:
- органов по контролю за лекарственными препаратами;
- органов по контролю за их качеством;
- производителей исходных веществ и лекарственных препаратов».
Истоки ЕФ относятся к 1964 году, когда шесть стран – членов Совета Европы подписали Конвенцию по разработке ЕФ. В настоящее время к ней присоединились все страны ЕС, а также многие европейские страны, не входящие в ЕС.
Монографии и требования ЕФ имеют определяющее значение не только для стран ЕС, а также для всей Европы и ряда неевропейских стран. Многие монографии воспроизведены в национальных фармакопеях европейских стран, например Британской фармакопеи (British Pharmacopoeia), Германской Фармакопеи (Deutsches Arzneibuch), Французской Фармакопеи (Pharmacopée Française), Итальянской Фармакопеи (Farmacopoeia Ufficialle della Republica Italiana), Государственной Фармакопеи Украины (Державна Фармакопея Украïни) и др. Часто требования, регламентируемые национальными фармакопеями стран-участниц Конвенции, принимаются в странах, исторически связанных с ними (например, Великобритания и страны Британского содружества).
ЕФ является обязательной в странах, подписавших Конвенцию, и к тому же приоритетной по отношению к национальным фармакопеям, что закреплено в Директивах ЕС, относящихся к лекарственным средствам (например, Директивы 65/65/ЕЕС, 75/318/ЕЕС и последующие документы).
ЕФ выдержала пять изданий, в настоящее время осуществляется подготовка ее шестого издания. Пятое издание ЕФ переведено на 32 языка. После 3-его издания выпуск последующих изданий осуществляется каждые пять лет.
Структура ЕФ включает общие статьи (General Chapters) и монографии (Monographs), определяющие требования к продуктам, предназначенным для применения у человека и в ветеринарии.
Общие статьи ЕФ содержат наиболее общие требования, распространяющиеся:
- на методы испытаний;
- на показатели (характеристики) качества;
- упаковочные материалы и контейнеры.
Сфера применения общих статей в последнем издании ЕФ существенно расширена. Это означает, что все лекарственные субстанции независимо от того, входят они или не входят в ЕФ, контролируются фармакопеей. Например, показатель «Остаточное количество органических растворителей», предусмотренный общей статьей ЕФ, должен определяться для всех субстанций.
Монографии ЕФ (общие и частные) регламентируют необходимые требования к показателям (характеристикам) качества и определяют методики испытаний, используемые для контроля качества:
- лекарственных субстанций (активных ингредиентов);
- вспомогательных веществ (эксципиентов);
- лекарственного растительного сырья;
- лекарственных препаратов, содержащих компоненты растительного происхождения;
- гомеопатических препаратов;
- радиофармацевтических препаратов;
- иммунобиологических препаратов.
Помимо указанных требований монографии содержат раздел «Производство», в которых изложены общие инструкции для производителей. Они касаются испытаний исходных веществ, технологического процесса, его валидации и контроля, испытаний готовой продукции, которые надлежит проводить производителю либо для каждой серии, либо для отдельных серий перед выпуском.
В отличие от Фармакопеи США (USP) в ЕФ не предусмотрены монографии на готовые лекарственные средства. В Директиву 75/318/ЕЕС внесены поправки о том, что при наличии общих монографий на лекарственные формы готовые лекарственные средства должны соответствовать их требованиям, а при отсутствии таких статей – требованиям национальных фармакопей государств ЕС.
Основной принцип ЕФ выражен следующим положением: «Качество продукта не отвечает ЕФ, если он не соответствует всем требованиям, указанным в монографии». Единство требований при оценке качества данного продукта независимо от исполнителя испытаний (производители, аккредитованные испытательные лаборатории) делает использование монографий ЕФ или ссылок на них идеальной основой для маркетинга лекарственных средств и вспомогательных веществ в масштабе мирового региона.
Другим преимуществом применения ЕФ является систематический пересмотр монографий, который осуществляется в каждом случае, когда требования монографии оказываются недостаточными для объективного контроля качества. Основанием для ревизии монографий являются следующие факторы:
- изменение качества продуктов на рынке;
- устаревание методик испытаний;
- изменение (например, ужесточение) требований к нормам отклонения показателей качества.
Применение новых технологии в фармацевтическом производстве может привести к появлению на рынке партий продуктов, характеризующихся иным профилем примесей, контроль которых в удовлетворительной степени не может быть выполнен в рамках действующей монографии. В этом случае изменение методик испытаний является вполне оправданным для объективизации контроля.
Замена устаревших методик испытаний происходит эволюционно в связи с прогрессом в развитии техники аналитического эксперимента. Новые возможности анализа способствовали внедрению концепции родственных веществ, сопутствующих в качестве примесей данному продукту. Если в более ранних монографиях тонкослойная хроматография была предпочтительным методом для их определения, то в настоящее время ведущая роль отводится методам газовой или жидкостной хроматографии. Немаловажно также и то, что последние позволяют проводить оценку не только содержания отдельных примесей (известных и неизвестных), но и суммы примесей, что само по себе представляет более жесткое требование.
Область применения монографий расширена за счет прилагаемого перечня известных и потенциальных примесей, которые могут контролироваться с помощью методик, приводимых в монографиях. Перечень содержит названия примесей и их структурные формулы и имеет исключительно информационный характер.
Разработка и пересмотр монографий ЕФ проводится в соответствии с «Техническим руководством для разработки монографий», принятым Европейской фармакопейной комиссией. Руководство представляет собой регулярно пересматриваемый и обновляемый общеевропейский стандарт по качеству монографий. Он содержит описание всех принципов, положенных в основу разработки монографий, которые могут быть использованы также национальными фармакопеями и производителями лекарственных средств.
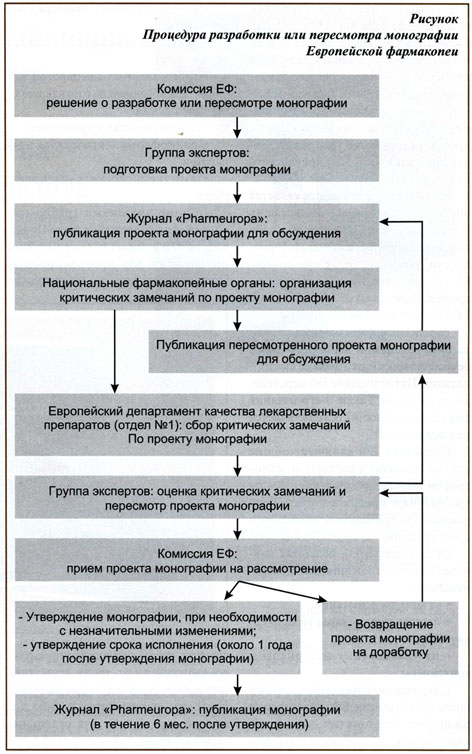
Процедура разработки и пересмотра монографий ЕФ представляет собой серьезный многоэтапный процесс, осуществляемый по схеме, изображенной на рисунке. Перед началом работы над очередным изданием ЕФ (в настоящий момент над 6-ым) обсуждаются вопросы целесообразности имеющихся общих статей и монографий, а также включения новых статей в фармакопею. Далее принимается решение о пересмотре статей в целях их обновления или разработке новых.
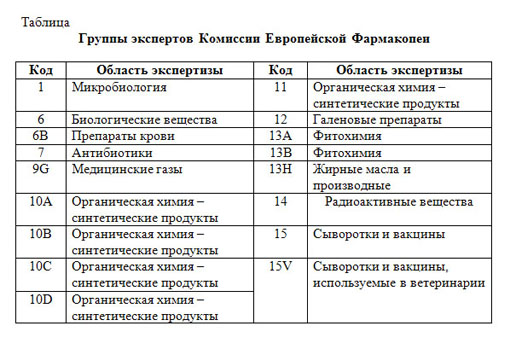
Следующим ключевым этапом схемы является подготовка проекта монографий, осуществляемая группами экспертов. В Комиссии ЕФ функционирует 17 групп экспертов, выполняющих экспертную работу в различных областях (табл.). Например, в области органической химии экспертизу проводит 5 групп, фитохимии – 2 группы, остальные разделы (антибиотики, медицинские газы, галеновые препараты, радиоактивные вещества, сыворотки и вакцины и др.) – по 1 группе. Этими же группами экспертов проводится оценка критических замечаний, внесение изменений и окончательная доработка проекта монографий.
Рассмотрение проектов монографий и последующее их утверждение (или возвращение на доработку) осуществляется на сессиях Европейской фармакопейной комиссии. Последняя такая сессия проходила с участием авторов настоящей публикации, представлявших республику в качестве наблюдателей. Обычно председатель заседаний на сессиях избирается на период два года, после чего проводится очередная ротация. Ноябрьская сессия проводилась профессором Ханнингом Г. Кристенсеном и была для него завершающей на посту председателя. Как правило, заседания Европейской фармакопейной комиссией блестяще организованы и проходят в исключительно демократической обстановке. Рабочими языками сессии являются английский и французский. Синхронный перевод на другие языки, например, русский или немецкий, должны обеспечивать при необходимости сами участники сессии. Наблюдателей, впервые присутствующих на сессии, сердечно приветствуют и приглашают к обсуждению документов сессии.
Монографии, утвержденные на сессиях Европейской фармакопейной комиссии, публикуются в течение 6 месяцев с момента утверждения и включаются в Дополнения ЕФ, которые издаются три раза в год.
ЕФ находится в состоянии постоянного развития и совершенствования, расширяются и модифицируются функции ее подразделений. Важной вехой в развитии ЕФ явилась реорганизация секретариата Европейской фармакопейной комиссии в Европейский департамент качества лекарственных препаратов (EDQM), основные функции которого заключаются в следующем:
- издание ЕФ (в том числе на компакт-дисках);
- издание журнала «Pharmeuropa» (4 выпуска в год);
- биологическая стандартизация лекарственная средств;
- создание сети европейских официальных лабораторий по контролю качества лекарственных средств.
Итогом многогранной деятельности ЕФ явилось формирование комплексной системы, цель которой сводится к обеспечению качества лекарственных средств и сырья для их производства. Система ЕФ предоставляет:
- монографии для контроля качества лекарственных средств и сырья для их производства;
- справочно-информационные материалы, необходимые для испытаний;
- техническое руководство для разработки монографий;
- информацию о проектах монографий, находящихся в процессе разработки или пересмотра;
- систему сертификации соответствия субстанций монографиям ЕФ;
- сеть европейских официальных лабораторий по контролю качества лекарственных средств.
Позитивным явлением в развитии Европейской Фармакопеи является стремление к гармонизации ее требований с фармакопеями США и Японии. Этот процесс осуществляется в рамках Международной конференции по гармонизации технических требований к регистрации лекарственных cредств для человека (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH). Проекты монографий, рассматриваемые в целях гармонизации, публикуются для обсуждения в органах печати фармакопей этих стран – «Pharmeuropa» в Европе, «Pharmacopeial Forum» в США и «Japanese Pharmacopeial Forum» в Японии.
Европейский опыт работы в области контроля качества и методов анализа лекарственных средств имеет важное значение в определении национальных подходов и путей развития в данной области. Для Казахстана это означает возможность его использования в разработке государственных стандартов и положений, устанавливающих требования к качеству и безопасности лекарственных средств, исходных веществ и вспомогательных материалов для их производства. Создание Государственной Фармакопеи Республики Казахстан, основанной на принципах, методах и методиках ЕФ, позволит:
- обеспечить высокое качество лекарственных средств, поступающих на рынок республики;
- стимулировать внедрение требований надлежащей производственной практики в отечественную фармацевтическую промышленность;
- защитить фармацевтический рынок от продукции сомнительного качества и различного рода фальсификации путем предоставления стандартов качества лекарственных средств;
- способствовать развитию научных исследований по созданию оригинальных лекарственных средств, в том числе из отечественного сырья.