

Вопросы экспертизы лекарственных средств государственной регистрации и оценка безопасности и качества зарегистрированных ЛС, ИМН и МТ
18.03.2013
 Наиболее часто встречающиеся ошибки заявителей при государственной регистрации и перерегистрации ЛС, внесении изменений в регистрационное досье были проанализированы в ходе встречи ведущих экспертов Национального центра экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники с представителями фармацевтических фирм. Встреча прошла под председательством А.Е. АБИЛАЕВА, генерального директора НЦЭЛС и вызвала большой интерес у менеджеров фармфирм, занимающихся подготовкой регистрационных досье.
Наиболее часто встречающиеся ошибки заявителей при государственной регистрации и перерегистрации ЛС, внесении изменений в регистрационное досье были проанализированы в ходе встречи ведущих экспертов Национального центра экспертизы лекарственных средств, изделий медицинского назначения и медицинской техники с представителями фармацевтических фирм. Встреча прошла под председательством А.Е. АБИЛАЕВА, генерального директора НЦЭЛС и вызвала большой интерес у менеджеров фармфирм, занимающихся подготовкой регистрационных досье.
Львиная доля рабочего времени и профессиональных усилий государственных экспертов тратится на «работу над ошибками», допущенными заявителями при подаче документов на регистрацию и перерегистрацию ЛС. Так, только 2% от общего количества заявок, поступивших на экспертные работы в 2012 г., прошло экспертизу «без сучка и задоринки», с выдачей положительных заключений. Об этом говорилось в докладе Алии КЕСИКОВОЙ, начальника управления первичной экспертизы лекарственных средств. Из приведенных ниже данных видно, насколько остра и актуальна тема.
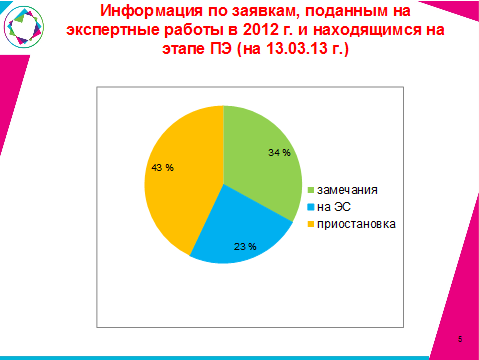
Информация по заявкам, поданным на экспертные работы в 2012 г., находящимся на этапе ПЭ ( по состоянию на 13.03.2013 г.)

Информация по заявкам, поступившим на экспертные работы в 2012 г.
По причине ненадлежащим образом составленных документов, предоставляемых на регистрацию, уже на этапе первичной экспертизы в 2012 году были отклонены 68% заявок, поданных фармфирмами стран дальнего зарубежья, 18% – СНГ и 14 % – Казахстана.
Как прокомментировал сложившуюся ситуацию А. Абилаев, «из-за некачественных регистрационных досье нашим сотрудникам приходится выполнять много ненужной работы». И это при том, что Центр в последние годы прилагает колоссальные усилия по созданию максимальной доступности, открытости и комфортности процесса государственной регистрации и перерегистрации ЛС.
Об этом свидетельствует и переход на работу по принципу «одного окна», и создание собственного интернет ресурса (dari.kz), на котором заявитель может познакомиться со стандартом госуслуги «Государственная регистрация, перерегистрация и внесение изменений в регистрационное досье лекарственных средств, изделий медицинского назначения и медицинской техники».
Здесь же представлена информация, какие документы нужны для сдачи регистрационного досье, образцы заявлений и много других полезных документов. У заявителя есть возможность, зайдя на сайт по логину, получить информацию о том, на какой стадии экспертных работ находится поданная им заявка. Если возникли проблемы, то можно задать вопросы руководству центра на сайте либо на еженедельных встречах, проводимых с заявителями.
Каковы же наиболее типичные недоработки при формировании регистрационных досье, с которыми сталкивается управление первичной экспертизы ЛС? О них Алия Кесикова рассказала весьма подробно на страницах журнала "Фармация Казахстана"(№3, 2013.г).
С проблемой недостаточной компетентности и непрофессионализма, некачественным исполнением своих функциональных обязанностей со стороны менеджеров фармкомпаний, занимающихся подготовкой документов для государственной регистрации ЛС, сталкиваются в повседневной деятельности и эксперты Фармакопейного центра. Ардак ТУЛЕГЕНОВА, начальник управления фармацевтической экспертизы НЦЭЛС, проанализировала все типовые несоответствия, которые встречаются на этом этапе проведения экспертизы.
Она отметила, во-первых, необоснованность отнесения заявленного ЛС к генерикам. Эксперт остановилась на таком аспекте, как возможность появления в воспроизведенных препаратах нового комплекса вещества или нового активного вещества, которые могут обладать совершенно иными свойствами. Такой препарат не может быть отнесен к генерику.
Во-вторых, необоснованность фармацевтической разработки. Необходимо обратить внимание на состав препарата, отсутствие взаимодействия между активным и вспомогательным веществами, отсутствие несовместимости физического, химического, фармацевтического характера.
То есть каждый вспомогательный ингредиент должен обладать именно теми функциями, которые установлены для него в Фармакопее. А сама лекарственная форма должна обеспечить высокий доступ активного вещества для максимального терапевтического эффекта.
Те же обязательные требования предъявляются к технологии производства и упаковке препарата. Что касается необоснованности спецификаций качества ЛС, то здесь зачастую встречаются погрешности, касающиеся данных по валидации. Предоставляемые на фармацевтическую экспертизу материалы страдают отсутствием или недостаточностью информации (Драг мастер-файл на субстанцию, фармацевтическая разработка, профиль растворения, стабильность, сертификаты анализа, валидация производственных процессов и прочее).
В ходе семинара был озвучен еще ряд замечаний, существенно мешающих проведению фармацевтической экспертизы. Это несоответствие АНД РК регистрационному досье (оригиналы на английском языке должны точно соответствовать АНД РК), несоответствие маркировки вторичной упаковки приказу МЗ РК от 08.06.2011 г № 366, несоответствие фармакопейным требованиям.
На сегодняшний день в Казахстане признаны пять Фармакопей, поэтому Ардак Уринбасаровна сочла нужным расставить приоритеты.
Первый уровень – Государственная Фармакопея РК. Именно с ее требованиями в первую очередь должны сверяться заявители. Но если в ГФ РК нет статьи по заявленному препарату, то следует руководствоваться Европейской Фармакопеей. Это уже второй уровень. К третьему отнесены Британская Фармакопея и Фармакопея США. Одновременное использование в одном регистрационном досье всех вышеперечисленных фармакопей недопустимо, предупреждает профессор Тулегенова, так как могут возникнуть разночтения.
Суммируя все замечания, докладчик обозначила основные причины отказа в регистрации со стороны фармацевтической экспертизы:
- Необоснованность состава ЛП.
- Качество, не соответствующее фармакопейным требованиям.
- Качество, худшее по сравнению с существующими на рынке Казахстана ЛС.
- Отрицательные результаты аналитической экспертизы по определяющим показателям качества ЛП и невоспроизводимость методик.
- Отсутствие ответов на замечания по истечении установленного срока (30 календарных дней).
- Недостоверность информации.
Особое внимание Ардак Уринбасаровна обратила на роль менеджеров фармкомпаний, занимающихся регистрацией препаратов. Сегодня большинство из них, по мнению эксперта, занимается «логистикой». То есть отвечают лишь за доставку документов в экспертный орган. Однако их главная задача – грамотная подготовка регистрационного досье.
Последние пять лет эксперты Фармакопейного центра сами проверяли соответствие предоставленных на регистрацию препаратов требованиям ФК РК. Пришло время четко определить зоны ответственности заявителя и эксперта. Перед тем как отправляться с документами в экспертный орган, заявитель (менеджер по регистрации или другое уполномоченное на это лицо) должен сам провести проверку соответствия регистрационного досье (АНД/ВАНД) фармакопейным требованиям.
В критических случаях, таких как несоответствие фармакопейным требованиям, недостаточность или несоответствие данных валидации аналитических методик установленным фармакопекйным требованиям, несоответствие данных испытания стабильности ЛС установленным законодательными актами требованиям, планируется оформлять отказ без этапа согласования.
|
Иной, более продуманный подход должен избавить экспертов от многомесячных малопродуктивных согласований, утомительной переписки и других обращений-воззваний к заявителям, высвободить время для рассмотрения добросовестно подготовленных регистрационных досье. Заявителям же предстоит пересмотреть отношение к этой сфере деятельности и всерьез взяться за повышение собственной квалификации. Для облегчения этого процесса составлен список литературы, размещенный на веб-ресурсе Центра. Там же можно прочитать доклады экспертов. |
На методах проведения специализированной фармакологической экспертизы и основных замечаниях этой службы к заявителям остановилась в своем сообщении Шынар БАЙДУЛЛАЕВА, руководитель фармакологического центра НЦЭЛС.
При проведении такого рода экспертизы эксперты руководствуются приказом МЗ РК №735. Согласно этого документа, при государственной регистрации требуется подтвердить безопасность и эффективность ЛС, к числу которых относятся:
- оригинальные препараты (п. 15);
- препараты-генерики (в зависимости от фармакологических свойств и лекарственной формы (п.п. 24-28);
- препараты-биосимиляры (п.23);
- гомеопатические лекарственные средства (п. 30);
- препараты растительного происхождения (п. 18);
- лекарственные средства с фиксированной комбинацией известных активных веществ, которые ранее не применялись в данном сочетании с терапевтической целью (п. 17);
- лекарственные средства, содержащие витамины и (или) представляющие собой комплекс витаминов и (или) витаминов и минералов (п. 29).
Обязательным является предоставление отчетов доклинических и клинических исследований ЛС, заявленных на регистрацию.
В отношении оригинального препарата основное замечание экспертов заключается в том, что заявители предоставляют только модуль 2, где дается общий отчет по качеству, обзоры клинических и доклинических данных и другая информация.
Данные для других модулей хранятся в головных офисах фирм-производителей, но не все менеджеры спешат запросить их и предоставить для экспертизы. Это, как отмечают эксперты фармакологического центра, существенно продлевает сроки проведения экспертизы.
Больше всего замечаний у экспертов при регистрации генериков (данные по ним предоставляются в модулях 4 и 5, в зависимости от фармакологических свойств и лекарственной формы). Все требования исходят из существующих международных рекомендаций доказательства биоэквивалентности препаратов. Минимальный уровень требований есть в рекомендациях ВОЗ, которым мы пока следуем.
В данном случае основные претензии к заявителям таковы:
- предоставление сокращенных отчетов исследований биоэквивалентности;
- отсутствие протоколов исследований (одобренная версия) и валидации биоаналитических методик;
- хроматограммы добровольцев;
- фармакокинетические кривые;
- плохое качество копий документов;
- неполная/некорректная информация о тест-препарате и (или) референс-препарате (серия, срок годности, производитель, страна-производитель, торговое название препарата, некорректный выбор препарата-сравнения);
- данные о добровольцах, выбывших из исследования;
- статистический анализ, информация об исследователе, месте проведения, периоде проведения исследований.
Увы, столь же внушителен список замечаний к заявителям при исследовании терапевтической эквивалентности. Часть их повторяет приведенные ранее. Это неправильно составленные отчеты, неполная/некорректная информация о тест-препарате и/или референс-препарате (серия, срок годности, производитель, страна-производитель, некорректный выбор препарата-сравнения), статистический анализ с большими погрешностями.
В большинстве заявок нечетко указываются первичные и вторичные точки оценки эффективности, отсутствуют описания динамики изменений в лабораторных, клинических параметрах и других показателей, использованных для оценки эффективности (УЗИ, ЭКГ, велэргометрия и т.д.). Отсутствуют мониторинг ПД и результаты исследования, выводы и/или заключение, предоставляются недостоверные данные.
Не стали исключением и препараты-биосимиляры. В регистрационных досье на них отсутствуют или в неполном объеме предоставляются следующие данные:
- сравнительные характеристики производства, качества активных веществ, готового продукта биосимиляра и оригинального препарата (банк клеток, примеси, первичная, вторичная, третичная и четвертичная структуры молекул, молекулярной массы, вспомогательных веществ и др.);
- отчеты сравнительных доклинических исследований биосимиляра и оригинального препарата (инвитро, инвиво, на моделях);
- отчеты сравнительных клинических исследований безопасности и эффективности, иммуногенности биосимиляра и оригинального препарата;
- фармаконадзор и план управления рисками на заявляемый препарат для обеспечения надлежащего мониторинга побочный действий и оценки соотношения польза-риск в постмаркетинговом периоде.
В протоколах и отчетах доклинических и клинических исследований лекарственных средств (оригинальных препаратов, генериков, биосимиляров), предоставленных экспертам, отсутствует идентификация исследования (код исследования; недостоверны ссылки на регистры клинических исследований). Нет идентификации страниц, подписей исследователей и спонсоров, информации о месте проведения исследования, главном исследователе. Отсутствуют приложения к отчету, информация об Этической комиссии, выдавшей разрешение, и даже само разрешение, а также разрешение регуляторного органа.
Есть нарекания к составлению инструкции по медицинскому применению заявленного препарата. Вся указанная в ней информация, говорят эксперты ФЦ, должна соответствовать данным исследований, представленным в регистрационном досье, и общей характеристике препарата с датой последнего пересмотра (в случае ЛС производителей стран СНГ – утверждённой инструкции по медицинскому применению в стране-производителе). Никакой «самодеятельности» быть не должно.
Информация, изложенная в проекте инструкции по медицинскому применению ЛС, сверяется с информацией на сайтах регуляторных органов других стран: FDA USA (fda.gov), EMA (ema.europa.eu), Федерального государственного бюджетного учреждения РФ «Научный центр экспертизы средств медицинского применения» (regmed.ru) и др.
При перерегистрации препарата не предоставляются PSUR (делается запрос на этапе специализированной экспертизы). Эксперты отмечают плохое качество PSUR: он неинформативный, отсутствуют данные мониторинга ПД заявляемого препарата, подпись лица, ответственного за фармаконадзор, информация об изменении профиля безопасности, нет анализа соотношения польза-риск, выводов, заключений.
После регистрации препарата особое внимание, по мнению экспертов, необходимо обратить на выполнение владельцами регистрационных удостоверений требований нормативных правовых актов, регламентирующих постмаркетинговый надзор безопасности лекарственных средств в РК. Приказом МЗ РК №735 предусмотрено предоставление PSUR в установленные периоды нахождения препарата на рынке Казахстана, но, как правило, такая информация не предоставляется.
|
Настоящей проблемой в ходе проведения экспертизы являются письма о приостановке экспертных работ по инициативе заявителей (43% от всех поданных на регистрацию заявок). По мнению экспертов, такие письма должны быть, во-первых, с обоснованием причин, во-вторых, только за подписью главы представительства. |
«Оценка безопасности и качества лекарственных средств, изделий медицинского назначения» – такова тема сообшения Раушан ТУРЫСБЕКОВОЙ, начальника управления по оценке безопасности и качества ЛС и ИМН.
Оценке безопасности и качества подвергаются все ввозимые в Республику Казахстан и произведенные на ее территории лекарственные средства. Докладчик познакомила аудиторию с нормативными актами, регламентирующими эту процедуру.
Осуществляется она путем проведения:
- серийной оценки безопасности и качества лекарственных средств, в том числе иммунобиологических препаратов, произведенных в соответствии с требованиями Надлежащей производственной практики GMP (РК, ЕС, США);
- оценки безопасности и качества каждой серии (партии) лекарственных средств, произведенных не в соответствии с требованиями Надлежащей производственной практики GMP (РК, ЕС, США);
- оценки безопасности и качества каждой серии (партии) лекарственных средств, не прошедших серийной оценки безопасности и качества.
Что касается изделий медицинского назначения, то оценке безопасности и качества подлежат стерильные изделия медицинского назначения независимо от класса безопасности, изделия медицинского назначения класса безопасности 2б и 3, изделия медицинского назначения класса безопасности 1 и 2а, контактирующие с кровью и внутренними полостями организма.
Оценка безопасности и качества изделий медицинского назначения осуществляется путем проведения:
- серийной оценки безопасности и качества изделий медицинского назначения, произведенных в соответствии с требованиями ИСО EN 13485, GMP (РК, ЕС, США);
- оценки безопасности и качества каждой серии (партии) изделий медицинского назначения, произведенных не в соответствии с требованиями ИСО EN 13485, GMP (РК, ЕС, США);
- оценки безопасности и качества каждой серии (партии) изделий медицинского назначения, не прошедших серийной оценки безопасности и качества в соответствии с вышеприведенными пунктами.
Типичные несоответствия лекарственных средств и изделий медицинского назначения требованиям регистрационного досье и нормативным документам по стандартизации касаются упаковок и маркировки:
- В регистрационное досье не внесены изменения в макете упаковки регистрационного номера лекарственного средства в виде обозначения «РК-ЛС-», что предписывается пунктом 8 постановления Правительства Республики Казахстан от 30 декабря 2011 года № 1692 «Об утверждении Правил маркировки лекарственных средств, изделий медицинского назначения и медицинской техники».
- Несоответствие упаковки требованиям нормативного документа (по дизайну, объему и др.).
- Несоответствие маркировки образцов ЛС и ИМН, утвержденным макетам упаковки (отсутствие маркировки на государственном языке, размещение надписей, картинок, переклеивание этикеток, несоответствующее качество упаковочного материала и др.).
В случае перерегистрации или внесения изменений в регистрационное досье по маркировке и упаковке лекарственного средства, говорят эксперты НЦ, разрешается ввоз в ранее утвержденной упаковке до 6 месяцев после внесения изменений по согласованию с уполномоченным органом. Допускается одновременная реализация лекарственного средства в ранее и вновь утвержденной упаковке до окончания срока годности лекарственного средства в ранее утвержденной упаковке (пункт 49 приказа МЗ РК от 15 февраля 2012 года № 84).
Далее встреча продолжилась в формате «Вопрос-ответ».
Один из вопросов – о количестве образцов лекарственных средств и изделий медицинского назначения, которые надо предоставлять для проведения испытаний. Этот вопрос, как пояснили эксперты, регламентируются Государственной Фармакопеей РК и нормативными документами по стандартизации.
Вопросов много, и они довольно актуальны для участников фармацевтического рынка. Об этом говорил Вячеслав ЛОКШИН, президент Ассоциации представительств иностранных фармацевтических компаний в РК, член коллегии МЗ РК.
Он озвучил весьма актуальную на сегодняшний день проблему. Дело в том, что в республику небольшими партиями (около 100 упаковок) стали завозиться дорогие препараты, стоимостью от 500 до 1000 евро. Для проведения экспертной проверки требуется 10-15 упаковок, что повышает стоимость ЛС для больного. Никакие объяснения, что на каждом из таких препаратов стоит электронный датчик температуры, а фирма-производитель гарантирует его сохранность и безопасность, не берутся во внимание.
По мнению руководителя одного из ведущих НПО, в таких случаях нужно искать компромиссный вариант, ведь нет правил без исключений.
В конце семинара Вячеслав Натанович от имени всех присутствующих поблагодарил руководство Центра за столь полезную инициативу.
Он высоко оценил предпринимаемые Национальным центром экспертизы ЛС, ИМН и МТ меры по переходу на международные критерии оценки безопасности и эффективности лекарственных препаратов, медицинской техники и изделий медицинского назначения.